ABC Heart Fail Cardiomyop 2025; 5(1): e20240054
Explorando o MYH7 nas Cardiomiopatias: Fatores Genéticos e Desfechos Clínicos
Lucas Vieira Lacerda Pires
![]() , Vinícius Machado Correia
, Vinícius Machado Correia
![]() , Layara Fernanda Vicente Pereira Lipari
, Layara Fernanda Vicente Pereira Lipari
![]() , Fernanda Almeida Andrade
, Fernanda Almeida Andrade
![]() , Fábio Fernandes
, Fábio Fernandes
![]() , Vagner Madrini Junior
, Vagner Madrini Junior
![]() , Mariana Lombardi Peres de Carvalho, Giovanna Napolitano, Elisangela Aparecida da Silva, Kelvin Henrique Vilalva
, Mariana Lombardi Peres de Carvalho, Giovanna Napolitano, Elisangela Aparecida da Silva, Kelvin Henrique Vilalva
![]() , Vitória Pelegrino do Val, José Eduardo Krieger
, Vitória Pelegrino do Val, José Eduardo Krieger
![]()
Resumo
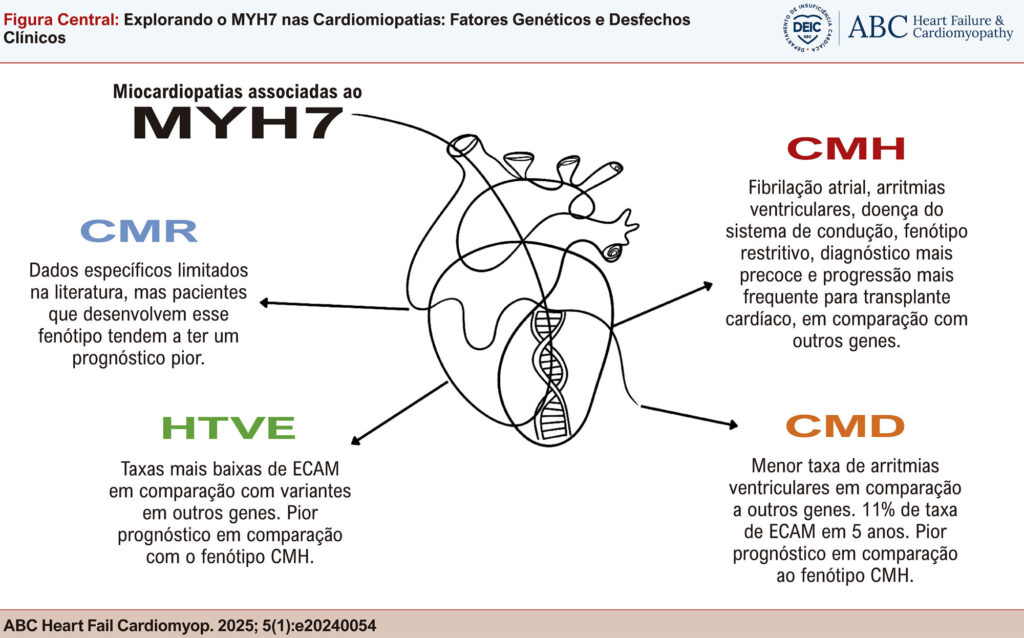
O gene MYH7, que codifica a cadeia pesada da beta-miosina, é um componente crítico na integridade estrutural e funcional das células musculares cardíacas e esqueléticas. Variantes no MYH7 estão entre as causas genéticas mais comuns de cardiomiopatias, particularmente cardiomiopatia hipertrófica (CMH) e cardiomiopatia dilatada (CMD), e também foram implicadas na cardiomiopatia restritiva (CMR) e com hipertrabeculação do ventrículo esquerdo (HTVE). Esta revisão explora os mecanismos moleculares pelos quais as variantes do MYH7 levam a esses fenótipos diversos, com foco nas correlações genótipo-fenótipo que fundamentam as manifestações clínicas de cada condição. As variantes do MYH7 são principalmente do tipo missense concentradas no domínio da cabeça da miosina, afetando a função contrátil da proteína. Essas variantes levam a um amplo espectro de anormalidades cardíacas, desde o espessamento das paredes miocárdicas até a dilatação das câmaras cardíacas. A revisão também aborda as implicações mais amplas das mutações do MYH7, incluindo seu papel nas miopatias esqueléticas e possíveis associações com o câncer. Compreender os mecanismos patogênicos das variantes do MYH7 não apenas aumenta a precisão do diagnóstico, mas também embasa o desenvolvimento de terapias-alvo. À medida que a integração de conhecimentos genéticos na prática clínica continua a evoluir, o gene MYH7 permanece um ponto crucial para o avanço na gestão e no tratamento das cardiomiopatias, oferecendo aos pacientes esperança de melhores desfechos clínicos por meio da medicina de precisão.
1.082